算法工具-药物发现加速器

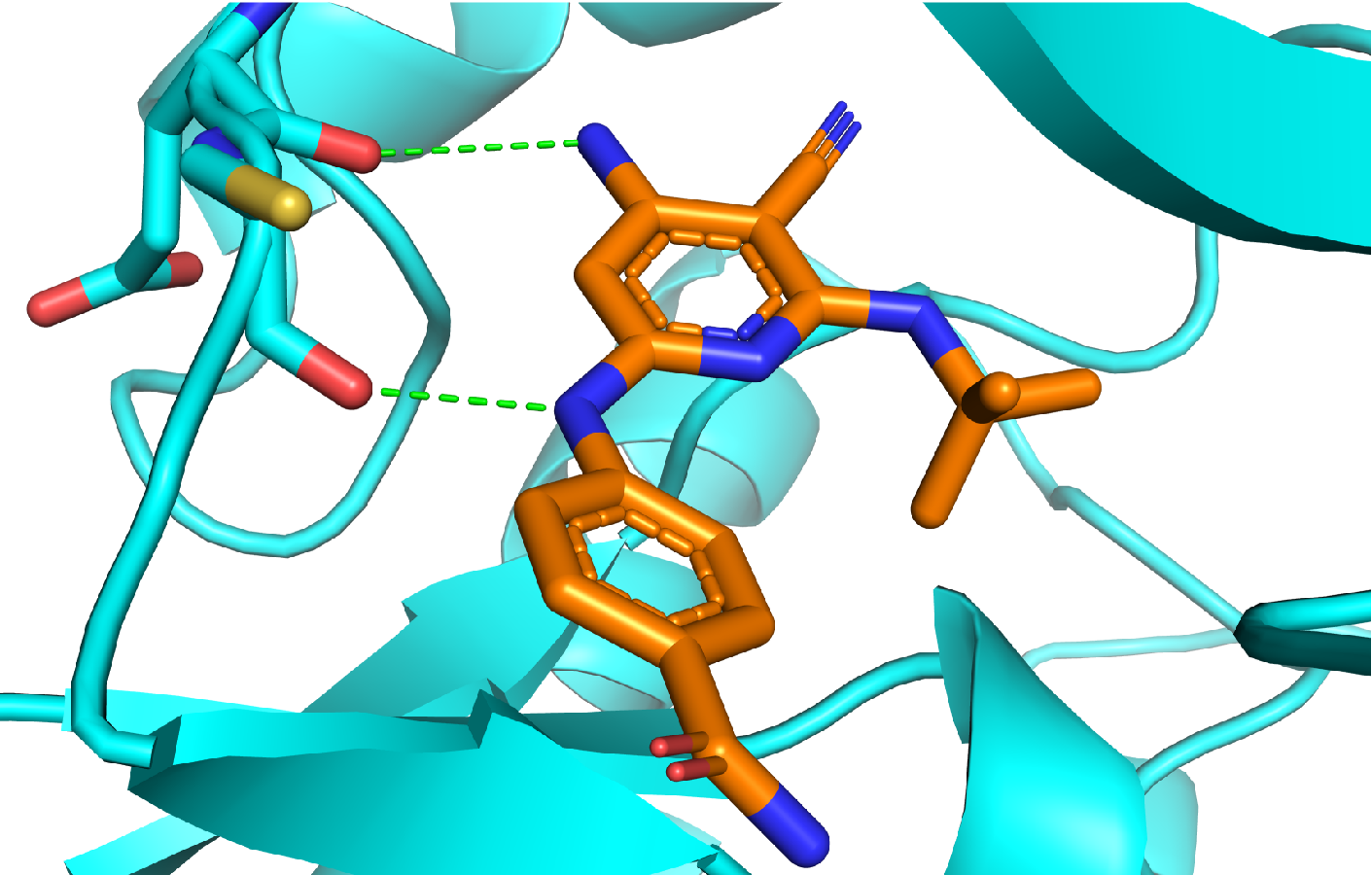
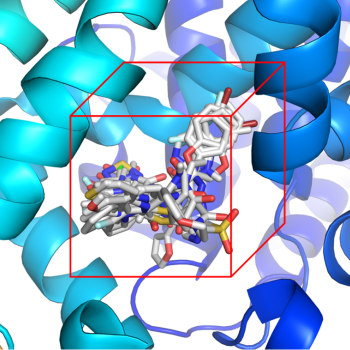
基于深度学习算法的蛋白-小分子复合物三维结构预测工具,根据三维结构可获得化合物与特定残基的相互作用信息,为小分子的结构优化提供线索。
分类 : 小分子&PROTAC对接工具
蛋白-小分子复合物结构预测可提供化合物与特定残基相互作用信息,对小分子的结构优化十分重要。
产品功能:
(1) 可根据用户上传的靶点蛋白质三维PDB结构文件,和提交的小分子结构文件,根据基于知识的打分函数预测并输出最佳的10个复合物结构,并给出初步的对接结合模式分析和结构显示。
(2) 对预测的复合物结构给出深度学习算法打分(可选)。
输入:受体蛋白质PDB结构文件,小分子结构文件(smi, sdf, mol2);
输出:最佳的10个复合物结构,经典打分文件,深度学习打分文件(可选)。
每次对接实验耗费10个点数。